Regulatorische Lösungen für Medizinprodukte und IVDs
Beratung und Unterstützung bei der Erstellung der technischen Dokumentation, beim Marktzugang sowie bei der regulatorischen Konformität von Medizinprodukten (MD) und In-vitro-Diagnostika (IVD).

Die regulatorischen Anforderungen für Medizinprodukte und In-vitro-Diagnostika sind komplex und können für Hersteller, Importeure und Händler eine erhebliche Herausforderung darstellen. Ramboll bietet maßgeschneiderte Beratung, um Kunden bei der Erstellung der erforderlichen technischen Dokumentation (TD) für eine erfolgreiche Marktzulassung zu unterstützen und die Einhaltung aller relevanten Vorschriften und Gesetze über den gesamten Produktlebenszyklus hinweg sicherzustellen.
Unsere Unterstützung während des gesamten MD-Lebenszyklus
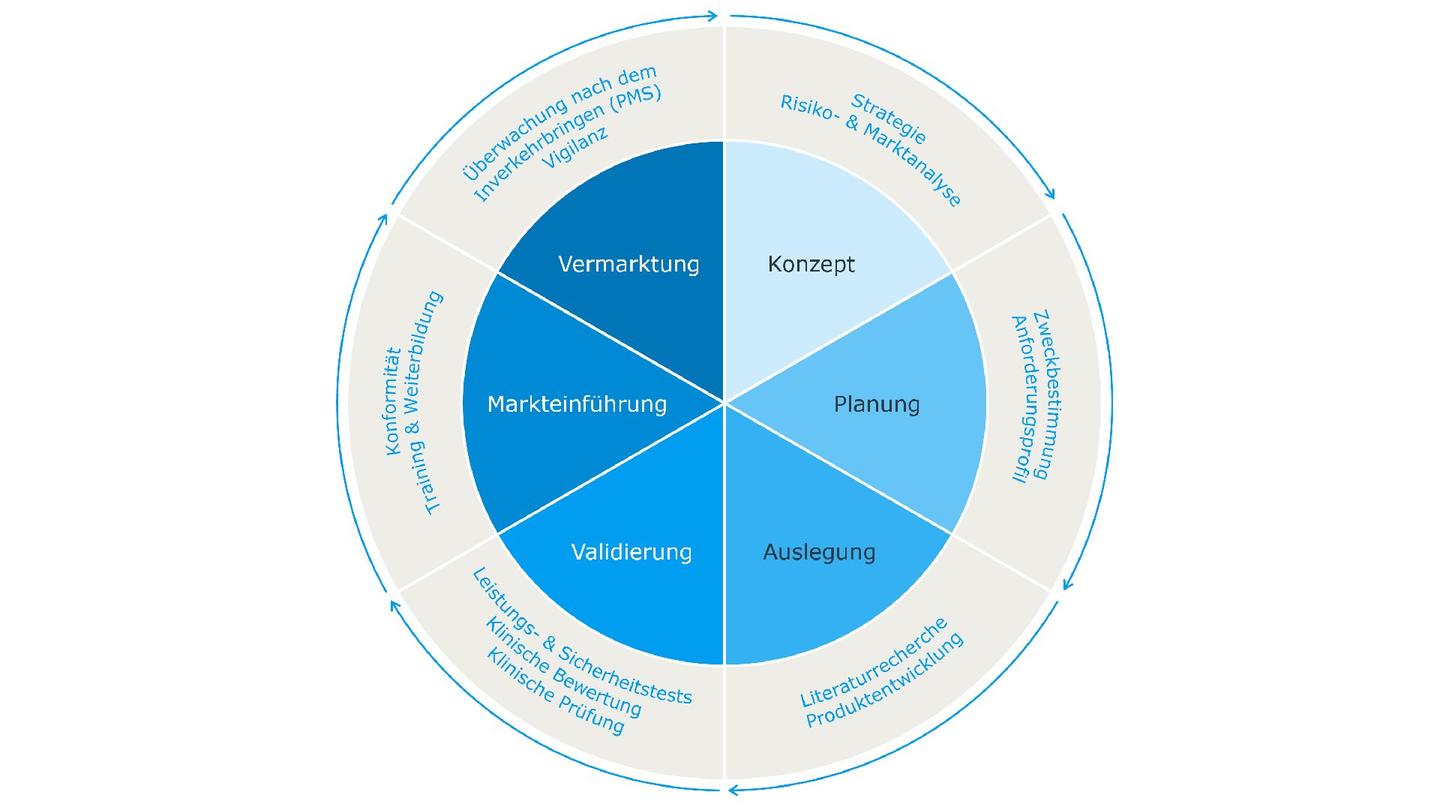
Mit der richtigen Unterstützung wird die Sicherung des Markteintritts und die Einhaltung regulatorischer Anforderungen deutlich einfacher. Wir entwickeln internationale Marktzugangsstrategien und bieten umfassende Unterstützung bei der technischen Dokumentation (TD).
Unsere Leistungen im Überblick:
Global konforme Qualitätsmanagementsysteme (QMS)
Die Implementierung eines robusten Qualitätsmanagementsystems (QMS) ist entscheidend für die Einhaltung der ISO 13485 sowie weiterer relevanter Standards und für Hersteller von Medizinprodukten verpflichtend.
Ramboll unterstützt bei:
- Erstellung und Pflege von QMS-Dokumentationen
- Entwicklung der Qualitätspolitik, einschließlich Planung und Definition der Ziele sowie der Festlegung von Leistungskennzahlen zur objektiven Bewertung von Prozessen
- Lückenanalysen sowie Recherchen zu Standards und Unterstützung bei internen und externen Audits
- Praktische Unterstützung bei Prozessoptimierung, Überwachung und Dokumentation
- Beratung zur EUDAMED
Risikomanagement nach ISO 14971
Das Verfahren zur Risikobewertung ist in der ISO 14971 beschrieben und wird in der ISO/TR 24971 weiter konkretisiert. Risikomanagement ist dabei ein kontinuierlicher, iterativer Prozess über den gesamten Produktlebenszyklus hinweg, der regelmäßige und systematische Aktualisierungen erfordert. Ein umfassendes Risikomanagement liefert Kunden wertvolle Erkenntnisse darüber, wie Medizinprodukte entwickelt werden können, die optimal auf die Bedürfnisse von Anwender:innen und Patient:innen abgestimmt sind.
Ramboll unterstützt bei:
- Entwicklung von Risikomanagementverfahren, die den regulatorischen Anforderungen entsprechen und international anerkannt sind, um Kunden bei der Reduzierung von Kosten und Zeitaufwand zu unterstützen
- Bereitstellung und Anpassung von Risikomanagement-Templates gemäß ISO 14971:2019 und ISO/TR 24971:2020 sowie Beratung bei deren Anwendung
- Schulung und Moderation von Risikobewertungs-Besprechungen
- Unterstützung bei spezifischen Fragestellungen und Abweichungen
Biokompatibilität gemäß der ISO 10993 Normen
Biologische Bewertungen und die Biokompatibilität von Medizinprodukten sind Risikomanagementaktivitäten, die nach ISO 10993-1 eine vorausschauende Planung erfordern. Ramboll begleitet Sie durch den gesamten Prozess – von der Erstellung des Plans für die biologische Beurteilung (BEP) über Labortests und die toxikologische Risikobewertung (TRA) bis hin zur Ausarbeitung des Abschlussberichts dr biologischen Beurteilung (BER).
Gemäß ISO 10993-1:2025 müssen Hersteller biologische Risiken über den gesamten Produktlebenszyklus hinweg bewerten, dabei vernünftigerweise vorhersehbare Fehlanwendungen berücksichtigen und aktualisierte Definitionen von Kontaktkategorien anwenden. Jede Bewertung beginnt mit der Analyse der verfügbaren Daten, einschließlich chemischer und physikalischer Informationen, biologischer Prüfungen sowie Daten aus der Überwachung nach dem Inverkehrbringen. Darauf aufbauend erfolgt eine maßgeschneiderte toxikologische Risikobewertung, die Unternehmen dabei unterstützt, Zeit und Kosten zu reduzieren, indem unnötige In-vivo- oder In-vitro-Tests vermieden werden.
Unsere Expertise umfasst:
- Risikobasierte Planung biologischer Beurteilungen gemäß ISO 10993-1:2025 sowie länderspezifischen Anforderungen (z. B. FDA-Vorgaben), unabhängig von bestehenden Laborkapazitäten
- Beauftragung und Überwachung chemischer Charakterisierungen (ISO 10993-18:2020 + Amd. 1:2022) sowie biologischer Prüfungen
- Datenbank-, Literatur- und in-silico-basierte toxikologische Risikobewertungen von extrahierbaren Substanzen gemäß ISO 10993-17:2023
- Erstellung biologischer Bewertungsberichte durch zertifizierte und registrierte Toxikolog:innen
Ramboll verfügt über umfangreiche Erfahrung in der Zusammenarbeit mit Laboren bei der Zusammenstellung von Prüfpaketen, darunter chemische Charakterisierung, Zytotoxizität, Sensibilisierung, Irritation, systemische Toxizität, lokale Effekte nach Gewebekontakt, Genotoxizität, Karzinogenität sowie Hämokompatibilität.
Darüber hinaus sind wir spezialisiert auf den Umgang mit besorgniserregenden Stoffen, Nanomaterialien und weiteren kritischen Substanzen wie per- und polyfluorierten Alkylsubstanzen (PFAS) sowie Mikroplastik. Wir bieten nachhaltige, strategische Lösungen, die über klassische Bewertungsansätze deutlich hinausgehen.
Klinische Bewertungen zur Einhaltung der EU-MDR
Hersteller von Medizinprodukten, die für den europäischen Markt bestimmt sind, sind verpflichtet, klinische Bewertungen gemäß Artikel 61 und Anhang XIV der EU-Medizinprodukteverordnung 2017/745 (EU-MDR) sowie unter Berücksichtigung mehrerer Leitlinien der Medical Device Coordination Group (MDCG) und der MEDDEV 2.7/1 Revision 4 Leitlinie durchzuführen.
Erfolgreiche klinische Bewertungen setzen eine präzise Definition des erforderlichen Umfangs klinischer Evidenz sowie die Auswahl geeigneter Leistungs- und Sicherheitsanforderungen voraus. Ramboll kombiniert hochrangige Konsensliteratur zum Stand der Technik mit umfassenden Recherchen klinischer Studien in Metadatenbanken, um sicherzustellen, dass alle relevanten Studien erfasst werden und die Bewertung auf der bestmöglichen klinischen Evidenz basiert.
Wir unterstützen bei:
- Erstellung oder Aktualisierung von klinischen Bewertungsplänen (CEP), Durchführung von Literaturrecherchen sowie Erstellung klinischer Bewertungsberichte (CER) einschließlich der klinischen Nachbeobachtung nach der Markteinführung (PMCF)
- Fachlicher Unterstützung bei internen Literaturrecherchen und der klinischen Bewertung
- Bearbeitung von Abweichungen, die von benannten Stellen festgestellt wurden
Sofern klinische Prüfungen erforderlich sind, unterstützt Ramboll zudem bei der Identifizierung eines geeigneten und kompetenten Partners.
Leistungsbewertungen für die Einhaltung der EU-IVDR
Für In-vitro-Diagnostika (IVDs) bieten wir Leistungsbewertungen gemäß Artikel 56 und Anhang XIII der EU-Verordnung über In-vitro-Diagnostika (IVDR) 2017/746 an, einschließlich der Leistungsüberwachung nach dem Inverkehrbringen (PMPF), um den Bedarf an Leistungsstudien für die CE-Kennzeichnung zu bestimmen.
Kontaktieren Sie Ramboll in Deutschland
Ramboll ist ein globales Architektur-, Ingenieur- und Beratungsunternehmen und deutschlandweit an mehreren Standorten vertreten.
Standorte in DeutschlandKontaktieren Sie unsere Expert:innen

Claudia Brakop
Senior Managing Consultant
Environment & Health

Dr Alexander Theis
Senior Managing Consultant
Environment & Health